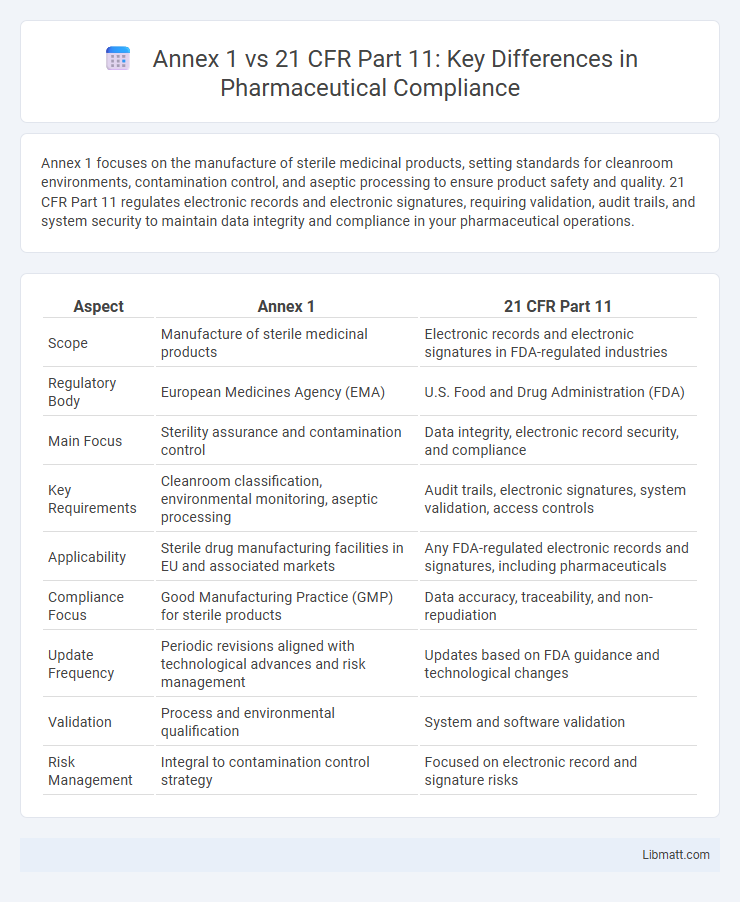
Annex 1 focuses on the manufacture of sterile medicinal products, setting standards for cleanroom environments, contamination control, and aseptic processing to ensure product safety and quality. 21 CFR Part 11 regulates electronic records and electronic signatures, requiring validation, audit trails, and system security to maintain data integrity and compliance in your pharmaceutical operations.
Table of Comparison
| Aspect | Annex 1 | 21 CFR Part 11 |
|---|---|---|
| Scope | Manufacture of sterile medicinal products | Electronic records and electronic signatures in FDA-regulated industries |
| Regulatory Body | European Medicines Agency (EMA) | U.S. Food and Drug Administration (FDA) |
| Main Focus | Sterility assurance and contamination control | Data integrity, electronic record security, and compliance |
| Key Requirements | Cleanroom classification, environmental monitoring, aseptic processing | Audit trails, electronic signatures, system validation, access controls |
| Applicability | Sterile drug manufacturing facilities in EU and associated markets | Any FDA-regulated electronic records and signatures, including pharmaceuticals |
| Compliance Focus | Good Manufacturing Practice (GMP) for sterile products | Data accuracy, traceability, and non-repudiation |
| Update Frequency | Periodic revisions aligned with technological advances and risk management | Updates based on FDA guidance and technological changes |
| Validation | Process and environmental qualification | System and software validation |
| Risk Management | Integral to contamination control strategy | Focused on electronic record and signature risks |
Overview of Annex 1 and 21 CFR Part 11
Annex 1 provides guidelines for the manufacture of sterile medicinal products focusing on aseptic processing and contamination control, ensuring product sterility and patient safety. 21 CFR Part 11 establishes criteria for electronic records and signatures in FDA-regulated industries, emphasizing data integrity, security, and audit trail requirements. Understanding both regulations helps you maintain compliance in pharmaceutical manufacturing and electronic documentation systems.
Historical Background and Regulatory Scope
Annex 1, issued by the European Medicines Agency, focuses on the manufacturing of sterile medicinal products and sets strict guidelines for aseptic processing and environmental controls. In contrast, 21 CFR Part 11, regulated by the FDA, addresses electronic records and electronic signatures, ensuring data integrity and security across all FDA-regulated industries. Understanding the historical development of Annex 1 since its initial release in 2008 and 21 CFR Part 11's establishment in 1997 helps clarify their distinct regulatory scopes and compliance requirements for your pharmaceutical operations.
Key Objectives of Annex 1
Annex 1 focuses on ensuring the sterility and safety of pharmaceutical products by providing guidelines for aseptic processing and manufacturing environments. It emphasizes contamination control, environmental monitoring, and validation of sterilization processes. The key objectives of Annex 1 are to minimize microbial, particulate, and pyrogen contamination risks to guarantee product quality and patient safety.
Core Principles of 21 CFR Part 11
21 CFR Part 11 centers on core principles such as ensuring the authenticity, integrity, and confidentiality of electronic records and signatures to comply with FDA regulations. It mandates secure user authentication, audit trails, system validation, and consistent record-keeping to prevent data tampering and maintain traceability. Understanding these principles empowers you to implement compliant electronic record systems that meet regulatory scrutiny effectively.
Document Management Requirements Comparison
Annex 1 and 21 CFR Part 11 both impose stringent document management requirements but differ in scope and application. Annex 1 emphasizes comprehensive control over documentation within pharmaceutical manufacturing, including paper and electronic records, ensuring data integrity, traceability, and audit trails. In contrast, 21 CFR Part 11 specifically governs electronic records and signatures, mandating secure user authentication, system validation, and detailed audit trails to comply with FDA regulations for electronic document management.
Electronic Records and Signatures: Compliance Factors
Annex 1 and 21 CFR Part 11 both establish critical compliance factors for electronic records and signatures in pharmaceutical manufacturing environments. Annex 1 emphasizes data integrity, security protocols, and validation requirements to ensure product sterility, while 21 CFR Part 11 focuses on audit trails, electronic signature authenticity, and system access controls to maintain regulatory compliance. Effective implementation of these standards demands robust software validation, user authentication mechanisms, and comprehensive documentation to meet global regulatory expectations.
Data Integrity in Annex 1 vs 21 CFR Part 11
Annex 1 emphasizes data integrity through strict documentation, traceability, and validation requirements to ensure sterility and compliance in pharmaceutical manufacturing. 21 CFR Part 11 mandates secure electronic records and signatures, focusing on system controls to maintain data accuracy, reliability, and audit trails. You must align processes with both regulations to guarantee comprehensive data integrity in pharmaceutical operations.
Impact on Pharmaceutical Manufacturing
Annex 1 and 21 CFR Part 11 significantly influence pharmaceutical manufacturing by enforcing stringent controls on aseptic processing and electronic records, respectively. Annex 1 emphasizes contamination control and sterile product integrity during manufacturing, while 21 CFR Part 11 mandates secure, compliant electronic record-keeping and signatures, enhancing data integrity and traceability. Together, these regulations drive robust quality management systems, regulatory compliance, and patient safety in pharmaceutical production.
Common Challenges in Implementation
Annex 1 and 21 CFR Part 11 share common implementation challenges, including ensuring data integrity, maintaining audit trails, and validating computerized systems. Companies often struggle with aligning compliance requirements with existing quality management systems, particularly in electronic record-keeping and electronic signature processes. Your organization must focus on robust documentation, user training, and regulatory risk assessments to navigate these challenges effectively.
Best Practices for Harmonized Compliance
Annex 1 and 21 CFR Part 11 establish critical frameworks for pharmaceutical and biotechnology industries, emphasizing sterile manufacturing and electronic records, respectively. Best practices for harmonized compliance include integrating validated electronic systems that ensure data integrity, audit trails, and secure user access aligned with Annex 1's contamination control requirements. Your approach should incorporate risk-based assessments and continual training programs to maintain regulatory alignment and quality system robustness across both standards.
Annex 1 vs 21 CFR Part 11 Infographic